|
The DNA sequences came back for the first round of specimens. As I understand it, to create the sequences a process similar to the PCR is done but the DNA nucleotides are exchanged for chain-terminating fluorescent nucleotides. That means each one of the four kinds of nucleotides in DNA is tagged with a specific color and when they are incorporated into the growing chain, DNA synthesis stops. The fragments of DNA are then sorted by size on a gel (using electricity to drive the negatively charged DNA towards the positive end). The machine then reads the different colors and converts the colors into their corresponding nucleotides. Ta-da you now have the sequence of nucleotides for that DNA. This is obviously the super simplified version and of course a lot of things can go wrong along the way. For a deeper dive into this topic you can read "Sanger Sequencing Steps & Method" by Millipore Sigma. 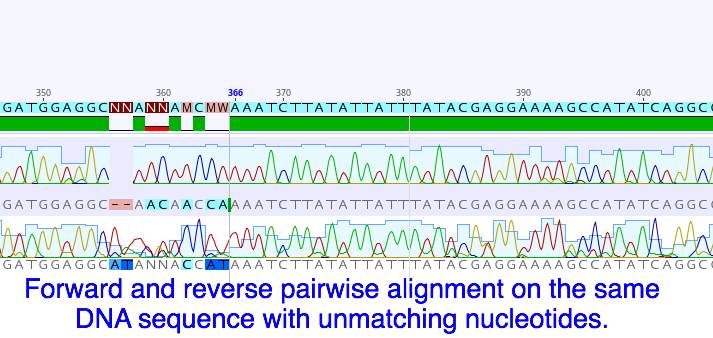 It is still possible the sequence will be unsuccessfully read. Even though we check the sample before it is sent out to make sure the DNA has been amplified, we have no way of knowing who's DNA was amplified. It is possible that the sample could get contaminated (don’t sneeze!) at some point during the extraction and amplification process. Another issue is the nucleotides in a sequence may not always come out clear enough to read with any certainty. This is why for each specimen we send a forward and reverse primer to initiate the sequencing reaction. This will give us two sets of the same sequence, one in each direction. Using a software program called Geneious we can overlap and compare the two sets for better accuracy. 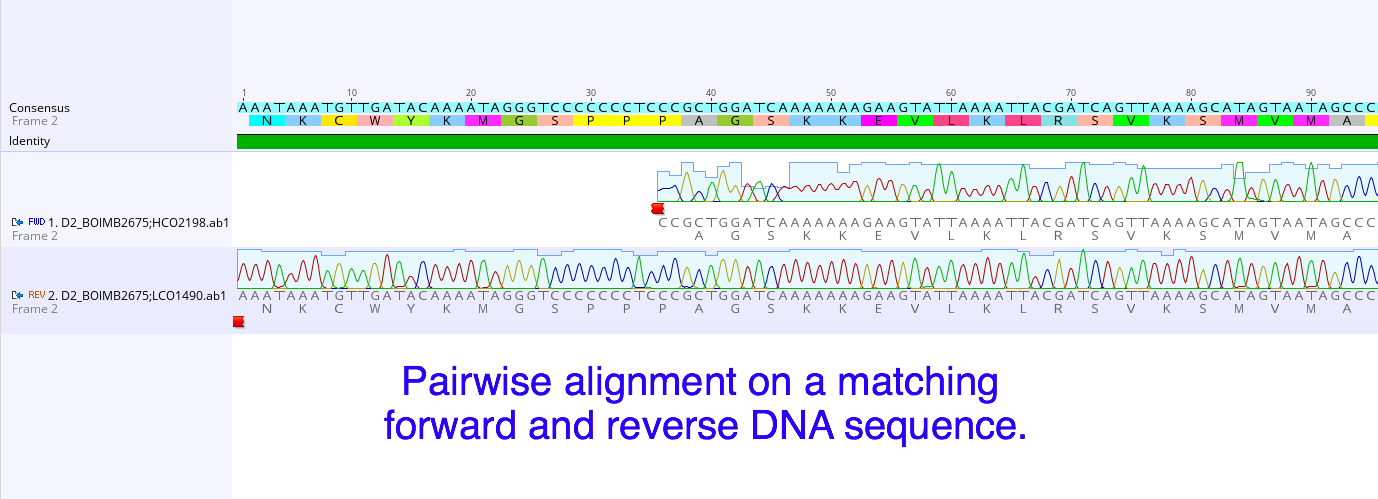 Working in Geneious has been really interesting. Once we trim up the two sequences, clean them up and merge them, we can then go on to the National Center for Biotechnology Information and upload the sequence to their program called the Basic Local Alignment Search Tool (BLAST). The BLAST program tracks uploaded sequences and their related species so we can use it to see if anyone has entered any information on known species that match our sequences. This part is exciting and really fun, most of the time it will also show you where the samples were collected from geographically. There is a lot more to this program and I’m looking forward to getting to know it over the next few weeks. Also this week we finished photographing and tagging all the worms we want to process from everything we’ve collected so far. We got all the DNA processed(extracted, amplified, and purified) and are hoping to ship out our second batch on Monday. In between that we have been able to look over and organize the BLAST information on the DNA sequences. The last round of field collecting should be this week also, which we’ll start processing as soon as we can. Hopefully we should have all the specimens processed and sequenced by the end of next week. Then we will be focusing on getting the information organized and displayed properly for the identification guide.
0 Comments
Leave a Reply. |
Rebecca OrrHi, my name is Rebecca I’m from Northern California. My major is in biological sciences. I am so excited to be working in Dr. Svetlana Maslakova’s lab and learning the protocol and procedures for completing a modern biodiversity survey. Archives
August 2021
Categories |
 RSS Feed
RSS Feed